互为等电子体,N₂O结构为什么和CO₂不同?
6 个回答

N2O,一氧化二氮(笑气)。


稍微思考不难写出其Lewis结构式如下。
N^{-}=N^{+}=O
//当然,有的人或许会更喜欢另一种共振式,即 N\equiv N^{+}-O^{-} ,我认为都是对的。
实验和理论计算结果都指出,N2O基态是单重态,为 C∞v 对称性的直线型分子,其中N-N键键长为1.127 Å,略长于氮气中的N-N三键键长。
利用现代计算机,我们也可以自己动手计算一下,r2SCAN3c的理论模型下,利用个人电脑,在Orca上几分钟就计算完成了,和文献结果对应很好。
根据计算结果,N2O的分子轨道排布可以很容易地写出,为
(1\sigma)^{2}(2\sigma)^{2}(3\sigma)^{2}(4\sigma)^{2}(5\sigma)^{2}(6\sigma)^{2}(1\pi)^{4}(7\sigma)^{2}(2\pi)^{4}
仔细对比一下就会发现,其和CO2的分子轨道是非常相似的,形状也很像,只是其中轨道能量发生了细微变化。
//CO2的为 (1\sigma)^{2}(2\sigma)^{2}(3\sigma)^{2}(4\sigma)^{2}(5\sigma)^{2}(6\sigma)^{2}(7\sigma)^{2}(1\pi)^{4}(2\pi)^{4}
进一步地,Mayer键级指出,N2O中,N-N键键级为2.29,N-O键键级为1.99,都非常接近2,这点与CO2也很类似。
当然,N2O与CO2不一样的地方也一眼可见,那就是其具有极性,非常显然,末端的O原子带有的负电荷会比末端N原子更多一些,同时中间的N原子会带有正电荷。
关于电荷,不同计算方法会给出不同结果,大体来说,N2O中电荷可以认为是如下分布(注意相对大小即可,不要过度解读数值绝对值)。


作为对比,CO2中的电荷分布大概是下面这样。


最后再补充一句,N2O中的这两个双键,和经典意义上的双键是不一样的(例如乙烯中的C=C双键)。
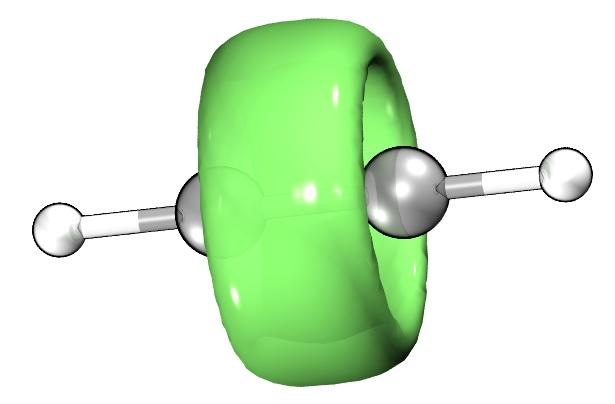
这点从N2O \pi 电子的ELF(Electron Localization Function, 电子定域化函数)图可以看出。


如上图所示,ELF函数展示的\pi 电子呈现出两个环形,这与乙烯的\pi 电子形状是非常不一样的,反而非常类似乙炔。
另外,电子明显偏向末端原子,说明极化明显,这和普通的共价双键(电子更倾向于在两个原子中间位置)也不一样。
//不严谨地,ELF图可以粗略认为体现了 电子云的分布。
//这里同时附上乙烯和乙炔的\pi 电子ELF图,供感兴趣的同学对比。



关于这个结果的进一步解读,可以进一步阅读如下内容。

不请自来。
两个答主都没有看懂题主的问题。
题主的问题的意思是,为什么都是 AB_2 通式的分子,但二氧化碳是碳在中间,两边对称,符合 D_{\infty h} 对称性,而一氧化二氮却是氧在一头,符合 C_{\infty v} 对称性。
这个问题其实是很有意思的。
一.共振论如何讨论这个问题
写路易斯结构的时候,我们可能会听到一个这样的话:摆分子骨架的时候,电负性大的原子一般不会在中间。
原因是比较简单的,因为如果电负性大的原子在中间,那么周围原子的电子全给中心原子拉过来了,那么中心原子周围电子就多了,就要出问题。
当然了这么说不大好,我们可以看一下共振结构式的情况。
按照正常的情况,一氧化二氮可以写出这种共振式:
N\equiv N^+-O^- (I式)\leftrightarrow N^-=N^+=O (II式)\leftrightarrow N^{2-}-N^+\equiv O^+ (III式)
按照共振结构式贡献的分析,显然三个共振式的贡献是I>II>>III,这符合和氧化亚氮分子有关的实验事实。
(I式里面虽然有电荷分离,但是负形式电荷在电负性大的氧上,II式则都在氮上,III式电负性大的氧带了正电,电负性小的氮带了过多的负电荷)
那么,如果氧在中间呢?
我们可以类似写一下,因为电子数一样,所以成键情况可以照搬上式,得到:
N^-=O^{2+}=N^-(IV) \leftrightarrow N\equiv O^{2+}-N^{2-} (V) \leftrightarrow N^{2-}-O^{2+}\equiv N (VI)
显然,我们发现了一个问题,IV-VI式里,中间那个氧明明电负性很大,却带上了大量的正形式电荷,而两头的氮电负性小,却带上了不少的负形式电荷,根据共振论的原则,IV-VI式显然是远不如I-III式稳定的。
再看看二氧化碳采取两种不同形式的情况:
O=C=O\leftrightarrow O^+\equiv C-O^-\leftrightarrow O^{-}-C\equiv O^+
O=O^{2+}=C^{2-}\leftrightarrow O^+\equiv O^{2+}-C^{3-}\leftrightarrow O^{-}-O^{2+}\equiv C^-
一对比就能看出来,二氧化碳为什么碳一定要在中间而不是两边。
二.分子轨道理论对于一氧化二氮结构的解释
开始解释一氧化二氮这个例子之前,我们要先看看一个直线型 AB_2 分子的分子轨道是怎么解释的。
我们先假定一下A和B是一样的原子。
要统一氧化亚氮的结构,我们可以从叠氮离子 N_3^- 出发,通过把其中的一个氮换成氧(电负性微扰论)来构建氧化亚氮的模型。
从直线型离子 C_3^{4-} 出发,把两个碳换成氧,就可以得到二氧化碳的结构。
先来看看第二周期直线型同核三原子分子的轨道如何处理。
先建立坐标系:

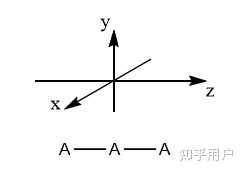
以分子的 C_\infty 主轴为z轴,同时建立垂直该轴的x,y轴,建立空间直角坐标系。
首先,考虑到 D_{\infty h} 点群,我们需要划定若干对称元素和对应的对称操作:
C_2(x) ,即x轴这个二重旋转轴,对应的对称操作是关于该轴旋转180°的操作
C_2(y) ,即y轴这个二重旋转轴,对应的对称操作是关于该轴旋转180°的操作
\sigma_h(xy) ,即xy平面这个对称面,对应的对称操作是关于该平面将某一轨道投射到该平面另一侧的对应位置上
i (对称中心),即上图坐标原点,中心A原子所在的点,对应的对称操作是把某一点关于该对称中心映射到相对应的另一点上。
至于为什么不考虑全等操作,因为没必要
至于为什么不考虑沿无穷次主轴的旋转,因为不能区分开除了σ型轨道以外的其它轨道
现在我们开始。首先,我们假定两侧的A原子的价层s轨道作为孤对电子不参与成键,那么两端的A原子只给出自己的三个价层p轨道。
因为是两个两个轨道去线性组合,只能是相加或者相减的形式(没挨着,没有重叠积分,不要忘记写波函数表达式的时候乘一个 \frac{\sqrt{2}}{2} 归一化),如图:


结合上面的坐标系去看,轨道符号的g,u表示这个轨道对于对称中心对称/反对称。
现在你可以老老实实去按照对称性匹配一个个分对称性,但如果是我会直接用肉眼去看。
可以想到的是, \sigma_g 轨道两端相位相等,指向中间的部分融合起来就会和s轨道差不多
\sigma_u 两端相位相反,指向中间的部分融合起来就和 p_z 差不多
\pi_u(x,y) 看起来就和 p_x/p_y 形状差不多
另外两个中心对称的轨道,看起来没有一个中心原子的轨道形状和它相匹配的。
于是,我们用于轨道组合的大体就是这么一些轨道了。
现在,我们来分析分子轨道.
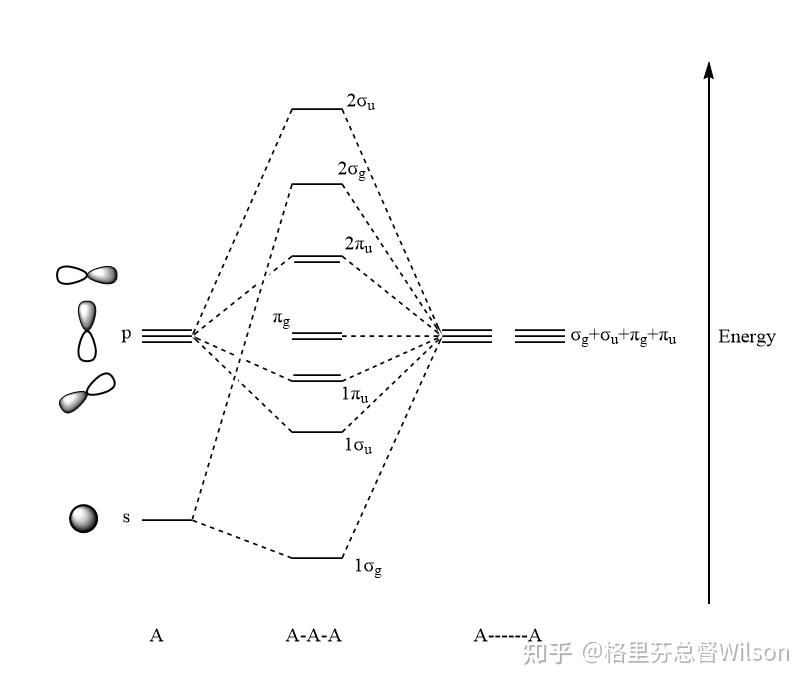
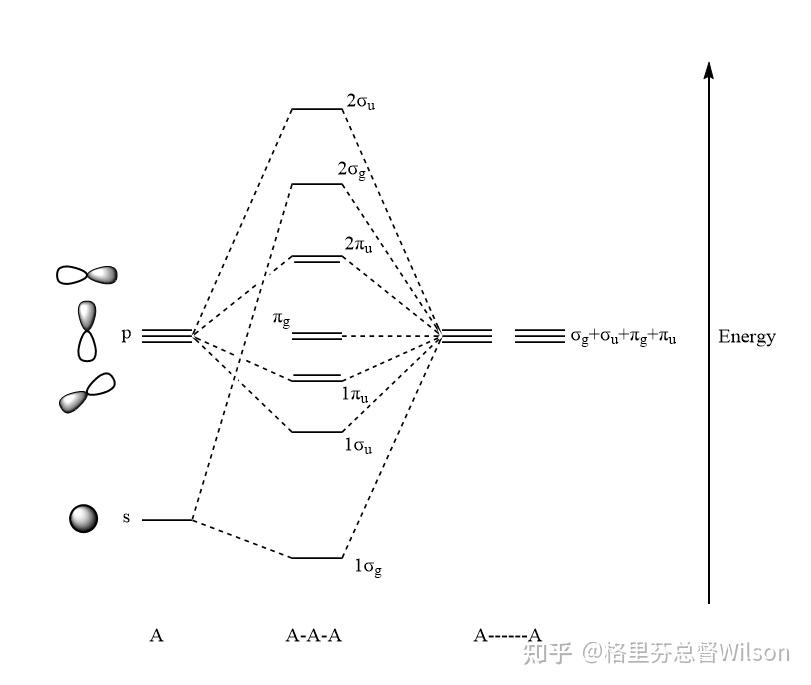
按照我们上面给出的对称性分析,把相同形状的轨道一线性组合,就可以得到上面的轨道能级图。
对于16电子的体系,以上分子轨道的电子分布式: (1σ_g)^2(1\sigma_u)^2(1\pi_u)^4(\pi_g)^4
现在我们来考虑一下置换的问题。
先考虑叠氮离子到氧化亚氮的变化,因为只换了一个轨道。
考虑到电负性微扰法的原理——
改变一个轨道的能量,所有占据轨道的能量会同时改变(此处是降低某一初始轨道的能量,所以所有相关分子轨道能量一起下降),如果原来系统的对称性被破坏,还会发生新的线性组合。
如果只把中间的氮换成氧的话,那么两个σ成键轨道和π成键轨道能量都要下降,除此之外没有什么明显的变化(对称性没变)
如果把一头的氮换成氧的话,那么所有的占据轨道能量都会发生下降,(这时π非键轨道也有氧的成分了)而且会导致新的问题——π成键轨道,非键轨道和反键轨道因为体系对称性的下降发生了重新组合,最后形成的三组π分子轨道里,会有一组能量更低的成键轨道,一组新的弱成键轨道和一组反键轨道(空的),这样就会使得体系的能量进一步下降,从而我们就可以解释一氧化二氮的氧在一头而不是中间了。
再看看从丙炔负离子 C_3^{4-} 到二氧化碳的微扰理论分析。
这个例子里面,如果把两边的碳换成氧,那么还是所有的占据分子轨道能量都要下降,而且因为对称性不改变,不存在新的线性组合。
如果把中间和一头的碳换成氧,那么还是所有的占据分子轨道能量都要下降,而且也会存在对称性下降导致的新的线性组合。
但是,因为氧的p轨道能量远低于碳,当两端的碳全部换成氧的时候,轨道能量变化是很明显的(尤其是只有端原子参与的非键轨道),而只把一端换成氧+轨道二次组合的能量下降是比不过前者的。
对于叠氮离子到氧化亚氮的变化,因为只换中间的氧不改变π非键轨道能量,而换端基的氧可以导致非键轨道能量的下降,所以自然氧就在端位。
三.总结
总结:三原子分子里面,电负性大的一定在末端而不是中间,所以氧化亚氮的氧是在一头而不是中间的,类似的氰酸根离子里面,最稳定的是 N^-=C=O,而不是 C^{2-}=N^+=O,更不可能是 N^-=O^{2+}=C^{2-}
其实我估计题主只想看共振论解释,甚至共振论都不想看,只想看经验规则。

不算回答问题。
我更好奇的是,为什么等电子体会相似?这个概念有什么细节吗?
我检索到的都是
These results are in contrast to their isoelectronic system
和
calculations of energy levels along the xxxxxx isoelectronic sequence
还有一些上年纪的物理评论
甚至是
Rayner-Canham, G. Isoelectronic series: a fundamental periodic property. Found Chem 11, 123–129 (2009). https://doi.org/10.1007/s10698-008-9055-4
但这并没有帮助

等电子原理是一个从大量的分子型化学物质,通过归纳的方法得出的,反映其“化学组成”(重原子数及价电子总数)与“结构特征”间关系的“分子结构规则”。是属于“现代价键理论”范畴内的一个“规则”。
这里的所谓“结构特征”,指的是“分子的环数”相等。如,C6H12这个分子式,可以代表许多种烃分子。它可以是双键位置不同的各种己烯,也可以是带支链或不带支链的环烷。但是,前者分子中有一个双键,后者则都有有一个“原子环”。如果把“双键”看作是一个二元的原子环,两者就“统一”起来了。也就是它们分子的“环数”都为“1”了。
等电子原理能告诉我们的,其实只是哪些等电子体的“环数”相等。教材中的所谓“结构相似”只是一个比喻性的模糊的说法。
更进一步的分析应该是:不管双键位置在何处的己烯,分子中都有两个碳原子为sp2杂化(其余4个碳原子为sp3杂化);而符合C6H12组成的环烷中所有碳原子都是sp3杂化的。
所以应该说,等电子原理可以确定的只是一个,受分子中“原子环”数目影响的,重原子杂化类型的某种“集合”。
这个原理根本就不涉及分子中重原子的排列顺序。如它对乙醇与二甲醚,在“结构特征”上是无法区分的(两者的化学组成相同)。它们的分子环数相同,还由于都没有“原子环”,所以两者的所有重原子杂化类型都相同(都是sp3杂化)。
至于,分子中原子的排列顺序,也就是分子的中心原子与配原子判别。教材中一般介绍到,要根据元素电负性。并规定“电负性小的元素居中”。
其实这也是一个不严密的说法。因为,它对于最为常见的H2O、NH3,就无法自圆其说。其实,还是要以原子半径与成键能力(可成共价键的数目),这两个因素来做标准才行。
原子半径大的,及可成共价键数目多的原子,一般才是中心原子。在N2O分子中,N的原子半径大,能成的共价键也多。所以N是中心原子。

这个问题问的好,别的答主也回答的特别好。
别的答主主要从理论回答,我从实验角度回答一波。
实验其实和理论还是有一些不一样的,实验中二氧化碳是特别不讲武德的一种气体。按照高中物理,碳和硅都是有四个价电子,但是常温常压二氧化碳是气体、二氧化硅是固体,是不是很神奇?
二氧化碳不能用简单的碳氧双键理解,我给你举个正常的东西做例子,叫做乙烯,乙烯才符合你理解的碳碳双键。乙烯常温常压是气体,加入催化剂进行加聚反应以后就变成聚乙烯了(碳碳双键断裂重新聚合),变成固体,是不是很神奇?
你看,二氧化碳常规催化剂加热加压的套路就不能自身加聚。所以说捏,你们书本上学的双键套路是不完备滴。二氧化碳你也不能简单理解为碳氧双键。
ok,以上答案主要解释二氧化碳为何化学性质和一氧化二氮不同,下边解释为啥结构不同。
你可能不理解为什么氧不能在中间对吧,因为实验中,氧本身它就是很难被别的元素抢电子,氮在中间让氧才侧边抢氮的电子才是符合大多数实验规律滴。
同理,氮气和一氧化碳也是等电子体,但是前者互不抢电子,后者氧“抢了”碳俩电子,两者化学性质天差地别。
总之实验中,化学键理论并不能很好的解释那些不讲武德的物质和现象。

先问是不是,再问为什么
这两个化合物的结构是相同的