硫酸根杂化类型是什么?
9 个回答

@想象中 @Waqira 跟着这两位逛知乎,今天无意发现大家对于硫酸根的杂化这类问题,居然还有这么大的争议,让我非常震惊。
如果连如此基础的一个问题都讲不清楚,无法达成一致意见,那么我们在科研中遇到的那些极其复杂的金属有机化合物,该怎么取得共识,该如何分析其碳-金属键的组成?
所以,这个问题不能回避,必须讲清楚。
//关于化学键的讨论可以非常非常深入和复杂,例如 N\equiv N 这么简单的一个分子,要是把其键长拉长一些,然后深入讨论,里面的门道就有很多,但是这属于前沿科学内容了。
对于 SO_{4}^{2-} 硫酸根这么简单的分子,我们有理由在基础教学领域取得共识。
以下是我的分析过程与结论。
首先从这个玩意是有晶体结构的,为正四面体,S-O键长几乎相等,且都短于S-O单键,这是我们的实验基础。
对于这么简单的分子,结构优化不用太高端的手段,用B3LYP/def2-SVP我都觉得有点杀鸡用牛刀了(但是反正现在计算机牛哔,几秒钟就算完了,无所谓了……)。
接下来,我们开始进行分析。
//以下分析都是用卢天博士开发的Multiwfn完成的。Multiwfn集成了很多实用工具,功能很强大,但是希望大家不要迷失在工具中,而要清楚明白自己想干什么,选择最正确的工具,而不是堆砌图画。
首先,Mayer键级分析指出,四根S-O键的键级都在1.45左右,介于单双键之间。
然后,我们对硫酸根做一个Pipek-Mezey定域化,这是一个非常有历史,但是被广泛认可的定域化方法,具体内涵自行查阅文献。
如下图所示,可以直观清晰地看到,O原子上,有两对p轨道上的孤对电子(轨道a与b),S-O之间有一根 \sigma 单键,最后O原子上还有一对s电子(轨道d)。
从轨道图一目了然,没有硫原子d轨道什么事情。


到这一步,其实做不做 @Waqira 的NBO分析都能得出结论了,那就是硫原子是sp3杂化,与每个氧原子成一根单键,然后每个氧原子上都有一个负电荷(这样才能具有两对孤对电子)。
这一点,从电荷分布也能看出来。
//不同划分电荷的方式,会导致电荷分布不一样,但是结论是定性一致的。
Hirshfeld原子电荷给出,S上带0.38个正电荷,O上带-0.59个负电荷(注意,这里数值的绝对值没有太大意义,只是用来定性衡量电荷分散程度),呈现很强的离子化特征。
所以至此我们也明白了,Mayer键级1.45中,1来自于S-O \sigma 单键,0.45来自于S正与O负的电荷吸引作用,全程没有什么d-p轨道交叠的事情。
以上就是关于硫酸根的分析。
为了更好的说明这个问题,我们再看一个例子,二甲亚砜(DMSO)。
如下到底哪个结构才是正确的?
教科书上画成S=O双键真的正确吗?


照猫画虎。
首先,Mayer键级分析指出,S-O键的键级在1.52,介于单双键之间。
然后,我们对DMSO做一个Pipek-Mezey定域化。
如下图所示,可以直观清晰地看到,O原子上,有两对p轨道上的孤对电子(轨道a与b),S-O之间有一根 \sigma 单键,最后O原子上还有一对s电子(轨道d)。


Hirshfeld原子电荷给出,S上带0.32个正电荷,O上带-0.40个负电荷(注意,这里数值的绝对值没有太大意义,是定性衡量电荷分散程度),呈现很强的离子化特征。
所以,DMSO中应该画成上图中电荷分离形式,而不是双键。同时也要看到DMSO中,电荷分离程度相较 SO_{4}^{2-} 而言,稍微好一点。
那是不是所有的极性双键都有问题?
不是的,我们看一个丙酮的例子。


照猫画虎。
首先,Mayer键级分析指出,C-O键的键级在2.18,是典型的双键!
然后,我们对丙酮做一个Pipek-Mezey定域化。
如下图所示,可以直观清晰地看到,O原子上,只有1对p轨道上的孤对电子(轨道a),C-O之间有一根 \sigma 单键(轨道c),还有一根非常非常明显的 \pi 键(轨道b)!
最后O原子上还有一对s电子(轨道d)。
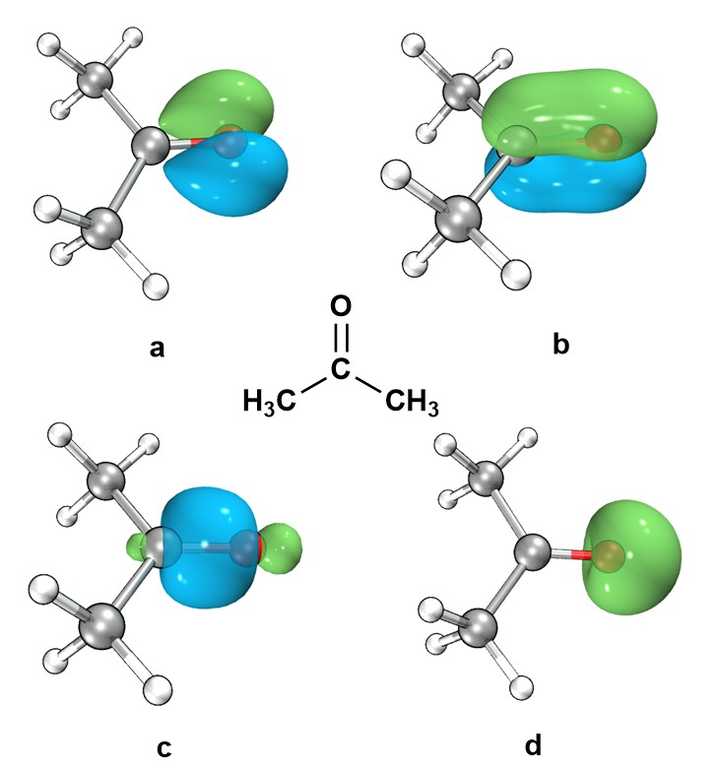

Hirshfeld原子电荷给出,C上带0.18个正电荷,O上带-0.25个负电荷(注意,这里数值的绝对值没有太大意义,是定性衡量电荷分散程度)。这就是根典型的极性键,不是离子键了。
所以,丙酮中应该画成上C=O双键。


跑题预警。中英文混杂预警。
在 @宮非 的评论区里提到我可以顺手用dft算了一下,那我就顺手用dft算了一下。
首先建议还是从点群入手。正四面体硫酸根为Td群,参考特征标表 [1]可以得到S原子上的轨道为:
a1+t2或a1+2 t2+e,如果要把d轨道也算进去的话。
之后再把4个O原子上的轨道组合起来,继续参考特征标表写出源于这4个O原子的16个轨道的对称性,然后再按照对称性和S原子上的轨道组合起来看看就知道了。至于d-pπ共轭什么的,其实我是不大信的。。。
总之,随便取了一个含硫酸根的玩意的晶体结构, [2]将其中的硫酸根部分取出来作为起始坐标,在ORCA 3.0.3 [3]上进行几何优化。使用def2-tzvp basis sets及def2-TZVP/J auxilliary basis sets、 [4] [5] [6]b3lyp functional, [7] [8] [9]同时引入d3 dispersion correction。 [10]将计算结果用NBO 7.0 [11]进行进一步分析,然后在Chemcraft [12]里将计算结果作图。
几何优化结果如下,键长与149pm [13]较为符合,就不进一步调basis sets和functional了。


试图分析分子轨道,发现我当年学的群论已经还给老师了,就不班门弄斧了。咱直接看NBO计算结果:


看Natural Charge那栏即可。NBO计算结果支持4个O原子等价的结论。注意Natural Charge和Formal Oxidation State(氧化态)不是一个东西。
其实这么看这很像一个S(2+)和4个O(1-)的结果。其中S丢掉两个电子,剩余4个电子分别和O原子成单键,然后每个O原子再额外获得一个电子,这样总体带-2的电荷,S和O也分别满足八隅体规则。
然后来看NBO计算得到的每个原子的电子结构:


emmmm S上还是有一小部分d电子参与的成分的。
然后每个原子的电子结构(如孤对电子blablabla的)直接跳过,分析成键。此处选取alpha轨道分析(alpha和beta一样的,大概是因为我用了uks算所以出来了alpha和beta轨道吧。。。),把alpha轨道电子数直接x2即可。
- S-O单键,以及S的杂化形式
具有高电子占据数的为4根S-O单键,基本等价。择其一而画。


对于该键的分析如下:


注意到首先是S-O键,键级为1【BD( 1)】。该键是极性键,其中S组分为32.62%,O组分为67.38%。其中S原子上成键轨道含24.99%的s轨道与72.86%的p轨道。
这是啥呢?差不多是个sp3杂化。
而O上轨道成分为24.73%的s轨道与74.65%的p轨道。这是不是sp3杂化呢?不是
原因在于O上只有一个p轨道和s轨道组合了,而且是不等性的。这两个轨道生成了一个低能量的轨道(上面放了一对孤对电子)和一个高能量的轨道(用来成键)。我们看一下O原子上的3个放孤对电子的轨道长啥样,及其成分就行了。


注意到最左边的那对孤对电子所在的轨道里有全部剩余的O s轨道成分,我个人觉得这玩意就别叫他杂化了吧。。因为其实这个s和p轨道混合一下生成两个新轨道的还蛮常见的。
然后再高一点能量的就是S-O反键轨道了,在此不表。
2. d-pπ共轭
注意到5个d轨道在td群里的特征标是不一样的(即写作e+t2),因此5个d轨道与参与的d-pπ共轭其实要分成两组,即dxy、dyz、dxz与dx2-y2、dz2要分开考虑。注意到此离子并非是平面四边形,因此有位答主给出的简图是错误的。
在四根S-O反键之后的5个轨道就是以S的3d成分为主了,我们姑且认为这5个轨道就是d-pπ共轭轨道。
在NBO计算里这5个d-pπ共轭的电子占据数极低,基本可以忽略的那种。注意到S-O单键的占据数为0.994/1=99.4%。而这几个d-pπ共轭里占据数较高的3个(t2)占据数在0.034~0.036/1=3.4~3.6%之间。较低的(e)甚至只有2%上下。
那么下面上图:
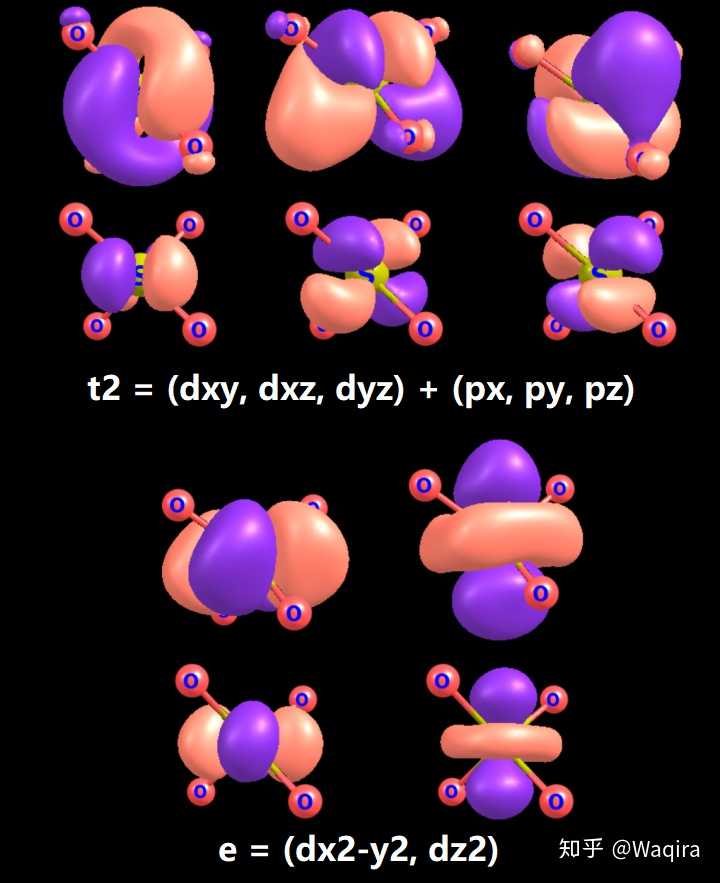

这什么玩意这能算有d-pπ共轭?????这就是S的3d轨道吧???而且dxy和dx2-y2你俩是不是串场了???
看一下这5个轨道的成分,一看便知:




好吧我还是觉得d-pπ共轭不怎么存在。。。
硬要画的话:


大概就是长这样的吧。
结论,我更倾向于S-O单键,S sp3杂化,O不杂化,但是s轨道与一个p轨道组合生成了一个低能量孤对电子+一个高能量成键轨道的解释。
但是注意到我这b3lyp-d3+NBO也只是一家之言。而且其实我对NBO不是特别熟,可能解读也有误。欢迎指正!
参考
- ^ http://symmetry.jacobs-university.de/cgi-bin/group.cgi?group=902&option=4
- ^ Eduardo Ernesto Castellano CCDC 1948978: Experimental Crystal Structure Determination, 2019, DOI: 10.5517/ccdc.csd.cc23f28x
- ^Neese, F. The ORCA program system. Wiley Interdisciplinary Reviews: Computational Molecular Science 2012, 2, 73– 78, DOI: 10.1002/wcms.81
- ^ Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571– 2577, DOI: 10.1063/1.463096
- ^ Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829– 5835, DOI: 10.1063/1.467146
- ^ Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297– 3305, DOI: 10.1039/b508541a
- ^ Becke, A. D. Density functional calculations of molecular bond energies. J. Chem. Phys. 1986, 84, 4524– 4529, DOI: 10.1063/1.450025
- ^ Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648– 5652, DOI: 10.1063/1.464913
- ^ Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785– 789, DOI: 10.1103/PhysRevB.37.785
- ^ Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456– 1465, DOI: 10.1002/jcc.21759
- ^NBO 7.0. E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis, and F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI (2018)
- ^Chemcraft - graphical software for visualization of quantum chemistry computations. https://www.chemcraftprog.com
- ^https://en.wikipedia.org/wiki/Sulfate

提供一点文献:
A quantitative definition of hypervalency
Marcus Durrant, Chem. Sci., 2015, 6, 6614







恭喜题主,这确实是个科研问题。现在XMVB已经开放免费获取了,呼唤各位基。。。。。。础化学知识爱好佬们,赶紧算一下试试。
@Waqira @魏俊年 @想象中


2020-11-27
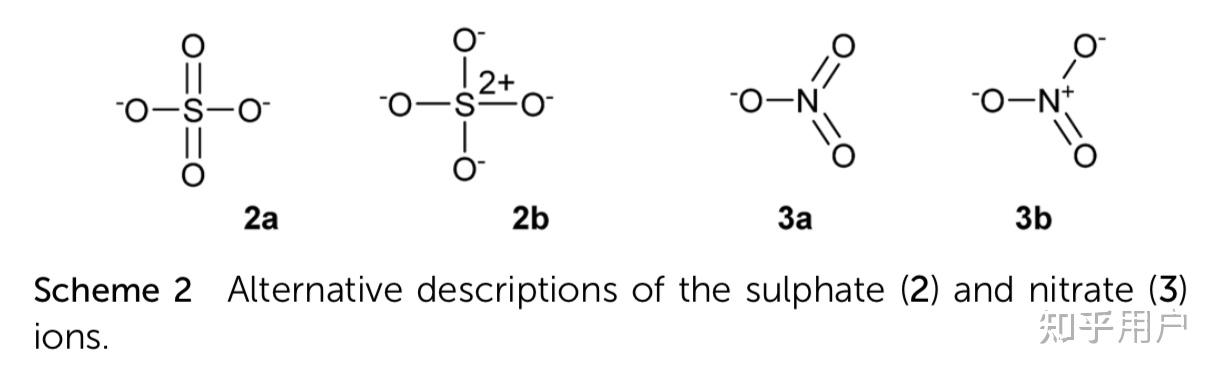
硫酸根离子中硫原子以 sp^3 杂化轨道成键,离子中存在 4 个 σ 键,很多说法称硫酸根是正四面体构型(见图 1),其实这是错误的,因为离子为「四面体型」(不是正四面体但接近正四面体,因为 4 个键的参数都不一样)。硫酸根中的氧原子的孤对电子和硫的 3d 轨道有 d-pπ 共轭效应,并非想像中那么简单(可能 5 组 d 轨道的形状本来就是有差别的),其结构至今在化学界没有定论,无法用一个单一的理论来解释。
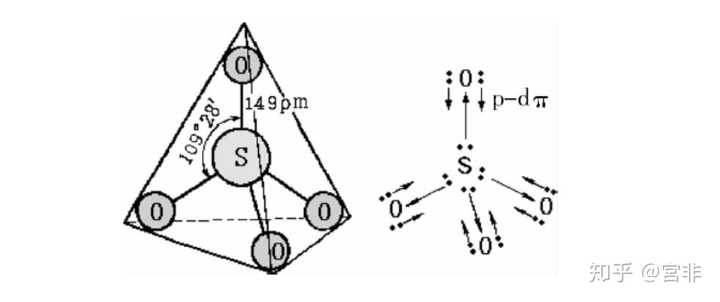

图 2 是经形式电荷计算后,硫酸根中各原子的形式电荷。


在图 2 中,每个氧原子的形式电荷皆为 −1,这没有问题,然而在高电负度的非金属硫原子上存在 +2 的形式电荷就显然不妥了,因此,需考虑其他可能的键结方式。若将氧原子上的孤电子对与硫原子的 3d 空轨域进行 pπ-dπ 键结,如图 3 之左图所示,硫原子的形式电荷为 0,这显然是一个较佳的结构。再者,与硫键结的四个氧原子条件皆同,以共振结构表示 S-O 的键结更合适,如图 3 之右图所示。实际测得的 S-O 键长为 149pm,比 S-O 单键键长 176pm 短了 27pm,可以确认 S-O 键不是单键,而应该是 1\frac{1}{2} 键。
注:S=O 键长为 143pm。


硫代硫酸根可看成 S0_4^{2-} 中的一个氧原子被硫原子所代替,并与 S0_4^{2-} 相似具有四面体构型(见图 4)。


分类:科普 >>化学 >>结构
参考北大出版«普通无机化学»,硫酸根sp3杂化,可以认为4个氧的p轨道与硫空的d轨道重叠,形成pi5,8大pi键。

硫酸根离子中,S原子采取sp³杂化,S原子上2个sp³杂化的孤对电子向2个端氧配体配位形成2个σ键,另外的2个单电子与另外两个氧负离子各形成一个σ键。
端氧的p轨道为了接受S原子的杂化轨道电子对,电子需要重排而空出一个p轨道。
端氧的p轨道与S的d轨道对称性一致,又会有端氧p轨道上的电子对向S原子空的d轨道配位形成d-pπ键。


常理上讲S应该是sp3
不过最近研究新键型的时候又有推翻的意味,具体内容忘了。高考sp3完事

中心原子是硫原子,杂化方式为sp3杂化,连接两个氧原子和两个羟基

听过一个说法不知道靠不靠谱,大多数分子的sigma键数加上孤电子对数就是杂化轨道数,这样来看硫酸根的硫应该是sp3杂化